Ремоделирование сердца
Содержание
 Термин «ремоделирование» вошел в медицинский лексикон в начале 80-х годов прошлого столетия. Вначале он был отнесен к сердечно-сосудистой системе — «ремоделирование сердца», «ремоделирование сосудов», а затем — и к другим структурно-функциональным образованиям.
Термин «ремоделирование» вошел в медицинский лексикон в начале 80-х годов прошлого столетия. Вначале он был отнесен к сердечно-сосудистой системе — «ремоделирование сердца», «ремоделирование сосудов», а затем — и к другим структурно-функциональным образованиям.
Интенсивное изучение процесса ремоделирования сердца (в основном, левого желудочка) началось после исследований SAVE (Международное многоцентровое рандомизированное двойное слепое исследование), установивших, что торможение процесса постинфаркгного ремоделирования левого желудочка сопровождается значительным улучшением течения и прогноза инфаркта миокарда
Что такое ремоделирование сердца
Согласно соглашению, принятому на Международном Форуме по Ремоделированию сердца в 2000 году (США), понятие «ремоделирование сердца» включает изменения на генетическом, молекулярном и клеточном уровнях, которые манифестируются изменениями структуры, размера, формы (архитектуры) и функции сердца, возникающими в ответ на долговременные повреждающие воздействия. К основные формам патологии, инициирующим ремоделирование сердца, относят ишемическую болезнь сердца, гипертоническую болезнь, гипертрофические кардиомиопатии и другие первичные заболевания сердца.
Одним из основных триггеров, запускающих процесс ремоделирования, является гибель кардиомиоцитов — их некроз (пассивная насильственная форма смерти), некроптоз (регулируемый некроз), апоптоз (активная программируемая смерть), аутофагия (смерть вследствие лиэосомального аутокатализа органелл, белков, липидов и других компонентов клетки). Некроз кардиомиоцитов сопровождается развитием асептической воспалительной реакции, при которой происходит активация транскрипционного ящерного фактора kappa В (NF-kB), детерминирующего синтез провоспалительных цитокинов, играющих ключевую роль в патогенезе многих процессов, включая ремоделирование сердца.
Ранее основными продуцентами провоспалительных цитокинов в поврежденном миокарде считали эндотелиоциты и тучные клетки. Затем было выявлено, что, кроме этих клеток, значительный вклад в процесс ремоделирования вносят фибробласты. Сравнительно недавно установлено, что эти клетки, кроме участия в пролиферации соединительной ткани, способны активировать инфламмасомы. Инфламмасомы (от лат. inflammatio- воспаление) — это цитоплазматические, формирующиеся в макрофагах и других клетках супрамолекулярные образования, которые способны опосредованно через стимуляцию каспазы-1 активизировать семейство интерлейкинов-1 (IL-la, IL-1J3, IL-IRa). В свою очередь стимуляцию кардиальных фибробластов могут вызывать активные формы кислорода — постоянные спутники ишемии, а также провоспалительные цитокины. Кроме того, ИЛ-la, ФНО-a, онкостатин-М и другие цитокины наряду с ангиотензином II, эндотели-ном 1 и катехоламинами активируют продукцию фибробластами матриксных металлопротеаз, которые являются членами семейства протеолитических ферментов, участвующих во многих биологических процессах.
Имеются основания считать, что матриксные металлопротеазы 3 и 9 принимают участие в процессе ремоделирования сердца. Контроль активности этих ферментов в значительной степени осуществляют тканевые ингибиторы матриксных металлопротеиназ — TIMPs (Tissue inhibitors of matrix metalloproteinases), которые образуют высокоаффинные комплексы с металлопротеазами, блокируя их активный домен, и таким образом предотвращают деградацию коллагена. К настоящему времени установлено, что преобладание активных матриксных металлопротеаз приводит к дилатации левого желудочка, а активная выработка TIMPs может способствовать его фиброзу.
Таким образом, в механизмах ремоделирования сердца участвует большой спектр кардиальных биологически активных молекул.
Геометрия левого желудочка изменяется на протяжении сердечного цикла от преимущественно эллипсоидной формы в систолу к более сферической в диастолу. Такие изменения закономерны в условиях нормальной насосной функции желудочка. Относительное удлинение левого желудочка во время систолы является механизмом, посредством которого желудочек выбрасывает больший объем крови при меньшем напряжении миокарда. Обратный процесс — сферификация левого желудочка во время ранней диастолы сопровождается увеличением объема желудочка и служит дополнением к раннему диастолическому наполнению, в котором участвует только пассивное удлинение кардиомиоцитов.
Два основных типа ремоделирования сердца
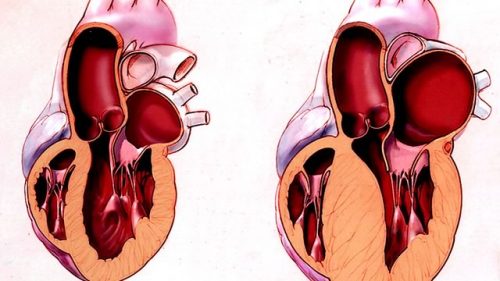
Выделяют два основных типа ремоделирования сердца: эксцентрический и концентрический (рис. 3.1). Критерием их дифференцировки является форма гипертрофии желудочка, которая представляет собой начальный этап ремоделирования. Тип ремоделирования детерминируется условиями, в которых он формируется. Например, объемная перегрузка левого желудочка при недостаточности аортального клапана вызывает увеличение длины кардиомиоцитов, уменьшение толщины стенок, увеличение объема и формирование эксцентрического типа гипертрофии левого желудочка. В отличие от такого типа ремоделирования перегрузка давлением левого желудочка (напр., в условиях стеноза аортального отверстия, системной артериальной гипертензии) приводит к увеличению количества саркомеров и объема кардиомиоцитов, толщины стенок и формированию концентрического типа гипертрофии левого желудочка.
В ходе изучения проблемы ремоделирования наряду с понятием «структурное ремоделирование» (изменения геометрии, архитектоники, объема, толщины стенки и т. п.) появилось понятие «функциональное ремоделирование». Применительно к насосной функции сердца функциональное ремоделирование ассоциируют с понятиями «систолическая и диастолическая дисфункция желудочка». Функциональное ремоделирование левого желудочка возникает и развивается независимо от процесса его структурно-геометрической перестройки. В настоящее время понятие «ремоделирование сердца» распространяется на все формы хронической сердечной недостаточности независимо от ее происхождения, т. е. от этиологических факторов.
На формирование и динамику процесса структурного ремоделирования сердца влияют гемодинамические, нейрогенные, гормональные и другие факторы, которые в настоящее время активно изучают.
При формировании концентрической гипертрофии повышение систолического давления стимулирует увеличение синтеза саркомеров в их параллельной ориентации, что вызывает нарастание массы миокарда и утолщение стенок желудочка, но не изменяет диаметр его полости.
При формировании эксцентрической гипертрофии повышение диастолического давления вызывает синтез последовательно располагающихся саркомеров. Для эксцентрической формы характерно увеличение массы желудочка и размеров его полости, но при этом средняя толщина стенок остается неизменной.
Гипертрофия левого желудочка закономерно развивается при артериальной гипертензии и способствует поддержанию напряжения его стенки. При этом развитие гипертрофии зависит не столько от уровня артериального давления (гемодинамической перегрузки), сколько от активности ренин-ангиотензин-альдостероновой системы. Вначале гипертрофия левого желудочка развивается по концентрическому типу (добавление саркомеров внутри кардиомиоцита). Ангиотензин II при этом стимулирует рост мышечных волокон, а альдостерон изменяет внутриклеточный матрикс с формированием диастолической дисфункции. Диастолическую дисфункцию, возникающую уже на начальном этапе ремоделирования левого желудочка, рассматривают в качестве маркера фиброза миокарда.
Расслабление миокарда
Расслабление миокарда — это весьма энерготребовательный процесс, и поэтому при гипертрофии желудочка он страдает в первую очередь. Наибольшую гемодинамическую перегрузку при диастолической дисфункции испытывает левое предсердие. Дилатация левого предсердия вызывает митральную регургитацию, что детерминирует переход концентрической гипертрофии левого желудочка в ее эксцентрическую форму. К систолической перегрузке повышенным артериальным давлением добавляется диастолическая перегрузка объемом, т.е. левый желудочек подвергается хронически повышенному конечно-диастолическому давлению. Дилатация левого желудочка осложняется систолической дисфункцией, что увеличивает риск летального исхода примерно на 50%.
При гистологическом исследовании стенки левого желудочка обнаружено повышение длины отдельных саркомеров и повышенное количество последовательно ориентированных саркомеров, которые, по видимому, приводят к увеличению длины миоцитов.
В процессе гипертрофии может развиться относительная коронарная недостаточность из-за отставания роста сосудов (ангиогенеза) от увеличения массы миокарда. Вследствие циркуляторной гипоксии и относительной недостаточности митохондриопоэза увеличивается количество необратимо поврежденных кардиомиоцитов, что приводит к снижению сократимости миокарда. В таких условиях кривая изоволюмического систолического давления продолжает смещаться вправо, а кривая диастолического давления может еще больше смещаться вниз (из-за фиброза), что детерминирует значительное снижение ударного объема и наступление у пациента терминальной стадии сердечной недостаточности.
Кардиомиоциты
Основными участниками процесса ремоделирования сердца являются кардиомиоциты, а также фибробласты и коронарные сосуды, а структурнофункциональной единицей сердечной мышечной ткани — сократительный (типичный) кардиомиоцит. Эти клетки образуют функциональные волокна, стыкуясь друг с другом. Места контактов соседних кардиомиоцитов называют вставочными дисками, которые свидетельствуют о клеточном строении миокарда.
Кардиомиоциты — это полностью созревшие, достигшие терминальной дифференцировки клетки, утратившие способность к делению в раннем постнатальном периоде. В связи с этим увеличение массы миокарда может обеспечиваться не образованием новых кардиомиоцитов и функциональных волокон, а лишь гипертрофией предсуществующих кардиомиоцитов. В ответ на возрастающую нагрузку кардиомиоциты не размножаются, а гипертрофируются — в них повышается синтез белка и саркоплазматических сократительных единиц. Индукторами гипертрофии являются норадреналин, ангиотензин И, эндотелии, локальные пептиды — стимуляторы роста клеток (инсулиноподобный фактор роста I, кардиотропин I, фактор роста фибробластов и др.), а также физические факторы, вызывающие растяжение кардиомиоцитов и повышение напряжения стенки полостей сердца. Взаимодействуя со специфическими рецепторами на мембране кардиомиоцитов, биологически активные индукторы запускают каскад внутриклеточных сигнальных цепочек.
В результате активируются гены раннего ответа (так называемые протоонкогены), ответственные за синтез малых регуляторных протеинов, контролирующих транскрипцию других генов. За этим следует реэкспрессия фетальной генной программы, которая, как показали экспериментальные исследования, индуцирует синтез сократительных белков и неконтрактиль-ных протеинов, таких, напр., как фермент р2 ~ Na+/K+-ATO-a3a, который обычно выявляют только у эмбрионов, т.е. в периоде, для которого характерна тотальная пролиферация клеток в организме. В отличие от других клеток, кардиомиоциты, остановленные в Gl-фазе клеточного цикла, способны отвечать на молекулярные стимулы лишь гипертрофией, но не пролиферацией.
В процессе ремоделирования активируется пролиферация фибробластов, что приводит к фиброзу — разрастанию соединительной ткани с появлением рубцовых изменений в сердце. Увеличивающаяся при этом «жесткость» миокарда детерминирует возникновение диастолической дисфункции, проявляющейся снижением насосной функции сердца.
Факторы регулирующие процесс ремоделирования
Симпатическая нервная система
Циркуляторная гипоксия, развивающаяся при сердечной недостаточности, вызывает активацию симпатической нервной системы, имеющую адаптивный характер и направленную на поддержание сердечного выброса (благодаря положительным хроно- и инотропным эффектам катехоламинов) и артериального давления. Однако пролонгация такой относительно несовершенной, т. к. используют лишь предуготовленные довольно лимитированные в своих возможностях компенсаторные механизмы, адаптации может придать ей патогенный характер из-за ее способности обусловливать прогрессирование ремоделирования и, таким образом, усугублять выраженность сердечной недостаточности.
Периферическая артериальная вазоконстрикция, наиболее выраженная в почках, висцеральных органах, коже и скелетных мышцах, направлена в основном на централизацию кровообращения, т. е. на сохранение притока крови к сердцу и головному мозгу.
Вазоконстрикция приводит к увеличению периферического сопротивления и, следовательно, постнагрузки на сердце. Вместе с тем, возможно также увеличение преднагрузки, т. к. при активации симпатической нервной системы повышается тонус венозных сосудов, что детерминирует увеличение притока крови к сердцу. Кроме того, неблагоприятные последствия пролонгированной активации симпатической нервной системы обусловлены повышением потребности миокарда в кислороде и энергетических субстратах, а также усилением в нем процессов перекисного окисления липидов (конечный продукт распада катехоламинов — ксантин является источником активных форм кислорода) и развитием проаритмогенного эффекта катехоламинов.
На более поздних этапах ремоделирования активированная симпатическая нервная система влияет на процессы реэкспрессии фетальных генов и гипертрофии кардиомиоцитов. В ряде исследований зафиксировано, что повышенный уровень циркулирующего норадреналина коррелирует с неблагоприятным долговременным прогнозом сердечной недостаточости у пациентов с дисфункцией левого желудочка, а использование (3-адреноблокаторов снижает летальность при данной форме патологии не только благодаря их антиаритмическому действию, но и способности тормозить процесс ремоделирования левого желудочка. Например, обнаружено, что (З-адреноблокатор метопролол может вызывать редукцию объема и регрессию массы левого желудочка, улучшая тем самым его геометрию.
Ренин-ангиотензин-алъдостероновая система
Спустя нескольких часов после начала развития острой сердечной недостаточности юкстагломеру-лярный аппарат (ЮГА) в почках увеличивает синтез ренина в ответ на снижение перфузии клубочков, точнее на снижение пульсового давления в v. afferens, которое отслеживают механорецепторы ЮГА, и активацию симпатико-адреналовой системы (в ЮГА имеются |32-адренорецепторы).
Таким образом включается ренин-ангиотензин-альдостероновая система (РААС). Современные научные данные свидетельствуют о параллельном функционировании гуморальной (циркулирующей) и тканевой (локальной) РААС. Локальные РААС функционируют в органах-мишенях, прежде всего в сердце, почках, мозге, сосудах, периферической мускулатуре. Ренин катализирует деградацию ангиотензиногена (относится к а2-глобулинам, синтезируется в печени) в гормон ангиотензин I, который затем при воздействии ангиотензин-превращающегося фермента (АПФ), находящегося в легких, почках и плазме, преобразуется в ангиотензин II.
Ген АПФ картирован в хромосоме 17q23. Различают две формы АПФ: мембранно-связанную (кининаза-2), которая обнаружена в макрофагах, Т-лимфоцитах, фибробластах; эпителиальных клетках почек, кишечника, плаценты, репродуктивных органах, и гуморальную (кининаза-1), которая образуется в различных тканях и органах, в основном, в эндотелии кровеносных сосудов легких.
В настоящее время установлено, что наряду с АПФ-зависимым механизмом преобразования ангиотензина I в ангиотензин II, существуют альтернативные пути — с участием химаз, катепсина G, тонина и других сериновых протеаз. Химазы, или химотрипсиноподобные протеазы, представляют собой гликопротеины с молекулярной массой около 30 000, обладают высокой специфичностью по отношению к ангиотензину.
В разных органах и тканях преобладают либо АПФ-зависимый, либо альтернативные пути образования ангиотензина И. Так, в ткани миокарда человека обнаружена кардиальная се-ринпротеаза. При этом доказано, что наибольшее количество данного фермента содержится в миокарде левого желудочка, где на долю химазного пути трансформации ангиотензину I приходится более 80%. Химазозависимое образование ангиотензина II превалирует также в миокардиальном интер-стиции, адвентиции и медии сосудов, тогда как АПФ-зависимое — в плазме крови.
Полагают, что активация альтернативных путей образования ангиотензина II играет большую роль в процессах сердечно-сосудистого ремоделирования. AT II является мощным вазоконстриктором, повышающим артериальное давление и стимулирующим секрецию альдостерона. Биологические эффекты ангиотензина II осуществляются через ряд рецепторов: I типа (имеются подтипы А и В) и II типа. Активация рецепторов 1 типа вызывает вазоконстрикцию и пролиферацию гладкомышечных клеток, а также стимуляцию процесса ремоделирования органов-мишеней.
На клеточном уровне AT II выступает в качестве индуктора синтеза трансформирующего фактора роста-р (ТФР-Р), который в свою очередь стимулирует хемотаксис макрофагов и фибробластов, индуцируя воспаление и активируя миофи-бробласты. Последние начинают синтезировать в избыточном количестве компоненты экстрацеллюлярного матрикса, что приводит к ускорению фиброзной перестройки сердечно-сосудистой системы. Структурные изменения стенок коронарных сосудов при воздействии AT II обусловлены пролиферацией гладкомышечных клеток и интерстициальных фибробластов, а также увеличением синтеза компонентов экстрацеллюлярного, соединительнотканного матрикса.
Из AT II образуется его метаболит АТШ, который обладает слабовыраженным прессорным свойством, но в значительной степени стимулирует секрецию альдостерона корой надпочечников. Альдо-стерон участвует в процессах задержки в организме ионов натрия, развитии вторичного гиперальдостеронизма и является фактором стабилизации АГ. Альдостерон обладает значительным профиброгенным эффектом, участвует в процессах ремоделирования левого желудочка сердца и сосудистой стенки, способствует развитию в органах-мишенях фиброза и функциональной недостаточности.
Антидиуретический гормон
Антидиуретический гормон (АДГ) — это пептид, состоящий из 9 аминокислотных остатков. У большинства млекопитающих, включая человека, в позиции 8 находится аргинин, такую форму АДГ обозначают, как аргинин-вазопрессин (AVP). Через VlA-рецепторы вазопрессин способен повышать сосудистый тонус. При физиологических концентрациях гормона его прессорный сосудистый эффект невелик.
В больших концентрациях АДГ вызывает спазмирование артериол, что приводит к увеличению АД и соответственно общего периферического сопротивления сосудов, отсюда название гормона — вазопрессин. Кроме того, АДГ поддерживает на должном уровне факультативную реабсорбцию воды в почках, уменьшая при этом диурез (антидиуретический эффект). АДГ образуется в супраоптическом и паравентрикулярном ядрах гипоталамуса, депонируется в задней доле гипофиза, откуда выделяется в кровь при возбуждении осморецепторов гипоталамуса.
При повышении осмотического давления плазмы АДГ выходит в кровь из нейрогипофиза. Способствуя реабсорбции воды в почечных канальцах, АДГ тем самым поддерживает венозный возврат к сердцу, т.е. его преднагрузку Такой эффект АДГ может иметь патогенное влияние на сердце в долгосрочной перспективе, особенно в условиях развития сердечной недостаточности.
К другим факторам, регулирующим процесс ремоделирования, относят натрийуретические пептиды, эндотелин 1, про воспалительные цитокины, оксид азота.
Натрийуретические пептиды
Различают три основных представителя семейства натрийуретических пептидов — предсердный, мозговой и С-конце-вой предсердный. При уменьшении сердечного выброса у больных с дисфункцией левого желудочка, а также при хронической сердечной недостаточности синтез натрийуретических пептидов увеличивается. Предсердный натрийуретический пептид выделяется в ответ на увеличение объема и давления в предсердиях. Мозговой натрийуретический пептид (тип В) образуется в головном мозге при растяжении его желудочков. Периферическая вазодилатация и натрийурез, вызываемые предсердным и мозговым натрийуретическими пептидами, противодействуют эффектам активации симпатической нервной системы и РААС, т.е. системной и почечной вазоконстрикции, задержке натрия и воды. В дополнение к их раннему благоприятному воздействию на гемодинамику, баланс жидкости и диурез, по данным некоторых экспериментальных исследований, долговременным эффектом натрийуретических пептидов может быть подавление гипертрофии кардиомиоцитов и. следовательно, создание благоприятных условий для «полезного» ремоделирования.
Эндотелии
Продуцентами этого пептидного гормона, представленного тремя изоформами, являются. Эндотелии является одним из сильнейших вазоконстрикторов; он гораздо более активен, чем ангиотензин II. Повышение уровня эндотелина в крови может быть причиной возникновения и утяжеления ишемической болезни сердца. В ряде исследований зафиксирован благоприятный результат блокады рецепторов к эндотелину у пациентов с сердечной недостаточностью. Не случайно поэтому эндотелии является маркером коронарного атеросклероза и эндотелиальной дисфункции коронарных сосудов.
В экспериментах установлено, что коррекция эндотелиальной дисфункции приводит к уменьшению массы миокарда левого желудочка, улучшению коронарной гемодинамики, увеличению силы сокращения миокарда, а также к подавлению синтеза внеклеточного матрикса фибробластов, что уменьшает степень выраженности периваскулярного фиброза венечных сосудов и предотвращает развитие интерстициального ремоделирования сердца.
Результаты исследования SOLVD (от англ., Studies of Left Nfentricular Dysfunction — Исследования дисфункции левого желудочка) подтвердили, что у пациентов с прогрессирующей сердечной недостаточностью повышается уровень провоспалительных цитокинов (ФНО-а, ИЛ-1, ИЛ-6 и др.), а за рубежом по критерию преимущественной «лекарственной терапии» XXI в. называют «веком иитокиновой терапии» В медицинском мире работы по изучению процесса ремоделирования продолжаются с перспективой использования их результатов для повышения эффективности патогенетической терапии пациентов с сердечно-сосудистой патологией.
Оценка ремоделирования
Установлено,что ремоделирование происходит на всех уровнях структурно-функциональной организации сердца и выражается в изменениях его размеров, формы и функциональных возможностей. Патофизиологический анализ и клиническую оценку ремоделирования левого желудочка проводят на основании измерения его линейных размеров и расчета ряда объемных показателей: индексов относительной толщины стенки, сферичности, миокардиального напряжения, нарушения желудочковой сократимости.
В настоящее время наиболее часто применяют методы определения геометрии и функциональных возможностей сердца: двухмерную эхокардиографию, магнитно-резонансную томографию и радионуклидную вентрикулографию. Необходимым условием динамического контроля за процессом ремоделирования является использование одного и того же метода в последовательных наблюдениях за состоянием левого желудочка каждого обследуемого пациента. Геометрия (архитектура) желудочка играет центральную роль в нормальной его функции и в процессе ремоделирования при различных заболеваниях сердечно-сосудистой системы.
При ухудшении насосной функции желудочков сердца увеличение преднагрузки направлено на под держание сердечного выброса. Длительная перегрузка инициирует ремоделирование левого желудочка: он становится более эллиптоидным, расширяется и гипертрофируется. Будучи изначально компенсаторными, эти изменения, которые иногда называют миокардиальным стрессом, со временем приводят к увеличению диастолической ригидности и напряжения стенки желудочка, что нарушает насосную функцию сердца, особенно во время физических нагрузок.
Увеличенное напряжение миокарда повышает потребность в макроэргах и при определенной степени развивающего дефицита энергии активирует апоптоз миокардиальных клеток. Итак, утрата нормальной эллипсоидной формы желудочка является ранним морфологическим признаком повреждения сердца, которая может стать пусковым стимулом развития хронической сердечной недостаточности.
Ремоделирование сердца предшествует клиническим проявлениям сердечной недостаточности и сопутствует им, т. к. способно усугублять систолическую и диастолическую дисфункции желудочка. На определенном этапе развития синдром ремоделированного сердца (другое его редкое название синдром «структурной кардиомиопатии») отодвигает на второй план значимость этиологического фактора, т.е. причины повреждения сердца, приводящего к развитию сердечной недостаточности.
На первый план выходит синдром «структурной кардиомиопатии» — патогенетический фактор сердечной недостаточности, определяющий механизмы ее развития, прогноз данной формы патологии и качество жизни больных. Изучение и понимание адаптивной и патогенетической роли ремоделирования сердца в каждом конкретном случае необходимо для избежания необоснованных терапевтических вмешательств, т.е. для оптимизации лечения пациентов с сердечно-сосудистой патологией.
 (No Ratings Yet)
(No Ratings Yet)
